
Retroperitoneal fibrosis (RPF) is a rare fibroinflammatory condition, causing fibrotic tissue deposit around the aorta, with potential extension to the retroperitoneum and surrounding structures.
Most frequently diagnosed in males (M:F 3:1) between 40-60 years old. RPF can be idiopathic or due to secondary causes [1-3].
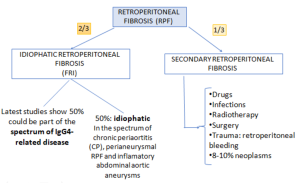
- Idiopathic retroperitoneal fibrosis is thought to be a multifactorial, immune-mediated disorder, most likely representing a manifestation of systemic autoimmune disease originating as primary aortitis. A cytokine-driven inflammatory cascade involving CD4+ T cells, IL-6, Th2 cytokines and TGF-β promotes B-cell maturation and IgG4-producing plasma cell expansion, leading to fibrosis. Genetic susceptibility appears limited, whereas smoking and asbestos exposure are recognised environmental risk factors.
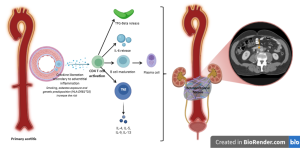
- Secondary RPF arises from medications, malignancy-related desmoplastic reactions, carcinoid-associated serotonin release, prior radiotherapy, or infection spreading from adjacent structures, though mechanistic pathways remain less well defined.
Clinical presentation is nonspecific, early and late symptoms depend on the time elapsed since onset [1,6,7].
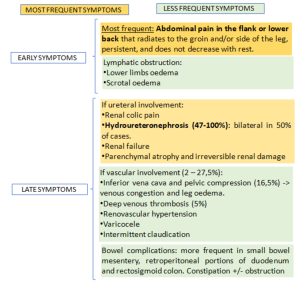
Although certain serologic markers may be used, and histopathology remains the gold standard for diagnosing RPF, its invasive nature has led to using less invasive approaches for both diagnosis and assessment of therapeutic response. Today's preferences are imaging techniques because despite imaging findings can overlap with other diseases, there are some characteristic findings which can favour RPF. Imaging may also help distinguish idiopathic from secondary causes and active from chronic disease [3].
