NF1 is an autosomal dominant hereditary disorder with an incidence of 1 in 2,000–2,500 live births. Approximately 50% of cases are inherited from an affected parent, while the remaining cases result from mutations in the NF1 gene, including microdeletions in the 17q11.2 region.
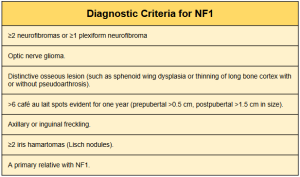
For clinical diagnosis, a set of recently revised criteria (2021) includes the presence of six or more café-au-lait spots, axillary freckles, bilateral inguinal freckles (ephelides), neurofibromas, optic nerve gliomas, Lisch nodules, bone dysplasias, a relative with NF1, and a heterozygous NF1 mutation. The presence of two or more of these criteria establishes the diagnosis of NF1.
One of the main characteristics of NF1 is the predisposition to developing benign and malignant neoplasms, including peripheral nerve sheath tumours (deep neurofibromas) and others such as brain tumours, phaeochromocytomas, paragangliomas, gastrointestinal stromal tumours (GIST), breast cancer, glomus tumours of the fingers (glomangiomas), and juvenile myelomonocytic leukaemia.
The risk of developing cancer in patients with NF1 is 59.6%, compared to 30.8% in the general population. The incidence of cancer development in children and women under 30 years old is particularly high in NF1.
Other characteristics of NF1 include alterations in cognitive development, such as autism spectrum disorder (ASD) and attention-deficit/hyperactivity disorder (ADHD), as well as other organic alterations such as aesthetic alterations, vascular pathology (Moyamoya disease), and skeletal defects.
This article, based on a series of cases from our hospital and a literature review, examines the imaging characteristics of PNF and the development of a standardised reporting approach. It also explores complications associated with PNF, including aesthetic and skeletal alterations, as well as its potential malignant transformation into malignant peripheral nerve sheath tumours (MPNSTs).