Radiological imaging is vital for assessing and following up on patients with beta thalassemia, enabling evaluation of both skeletal and extraosseous tissue involvement.
In some cases, clinical manifestations arise from severe chronic anemia that induces increased erythropoietin production. Erythropoietin stimulates the conversion of yellow marrow into hematopoietic red marrow in the bone. Medullary hyperplasia often leads to widening of medullary spaces, causing conglomeration and loss of normal trabecular structure. The expanding mass of red blood cell precursors can erode the bone cortex, compromising growth and causing skeletal anomalies. The severity of bone manifestations varies; remodeling can lead to osteopenia and severe cases may result in osteoporosis or pathological fractures.


All skeletal segments can be affected; however, areas with the highest hematopoietic activity—such as certain flat bones (e.g., ribs, sternum, cranial bones) and during youth, vertebrae and epiphyses of long bones—are most severely impacted. The skull may show an increase in diploic space with thinning of internal and external cortices, especially at the frontal bone level. Short bones may exhibit a radial appearance of trabeculae. Vertebral bodies can have a "grainy" or "ground glass" appearance with biconcave deformities at upper and lower boundaries. Reduction in vertebral body height is an early sign of severe osteopenia that can lead to vertebral collapse in young patients.Long bones may show cortical thinning, medullary dilation, and areas of osteoporosis. Phalanges may appear rectangular or biconvex—a non-pathognomonic feature also seen in other forms of tissue hypoxia.
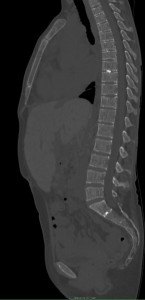
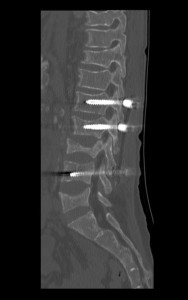
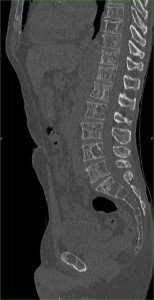
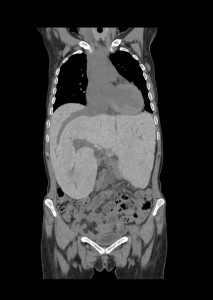
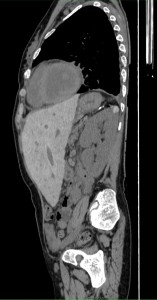
Despite appropriate therapy, marrow hyperactivity persists; extramedullary hematopoiesis can occur affecting the liver, spleen, and lymph nodes. In extreme cases, extraosseous masses can form in thorax, abdomen, and pelvis. These masses may compress adjacent structures causing skeletal deformities—particularly in the spine—which can result in neurological symptoms.
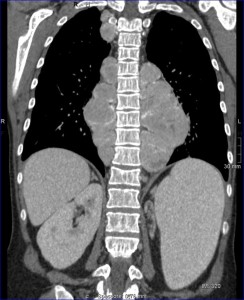
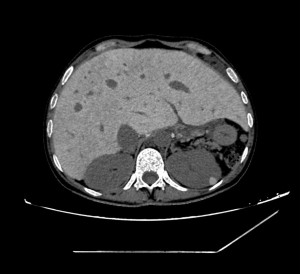
Splenomegaly is common and carries a risk of splenic rupture; prophylactic splenectomy may be necessary in cases with excessive hemocateresis. This can also lead to leukopenia and thrombocytopenia which reduce iron overload burden; however, levels remain elevated due to continuous blood transfusions despite chelation therapy.

Major consequences of iron overload include hypogonadism, hypothyroidism, hypoparathyroidism, diabetes mellitus, liver fibrosis, and cardiac dysfunction. Imaging plays a key role in diagnosing hepatic and cardiopulmonary complications.The liver typically appears hypertrophic with extensive fibrotic changes that may progress to cirrhosis and increase risk for HCV infection; severe cases may progress to hepatocellular carcinoma. Hepatomegaly is frequently associated with steatosis or cirrhosis.
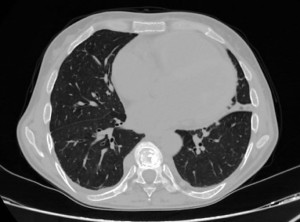
Common cardiopulmonary dysfunctions include restrictive pulmonary fibrosis visible on CT scans; impaired diffusing capacity for carbon monoxide (DLCO); small airway disease; and obstructive airway disease. Combined with excessive blood transfusions these factors can lead to pulmonary hypertension, cardiomegaly, and heart failure.
Cardiac complications are the leading cause of death among patients with beta-thalassemia major; thus follow-up with a multidisciplinary team is essential for monitoring cardiac health. The radiologist contributes to evaluating myocardial iron overload through cardiovascular magnetic resonance (CMR) using T2* imaging. Damage from excess iron manifests as irreversible dilated cardiomyopathy; myocarditis is also common due to anemic state and chronic inflammation making patients more prone to infections.